 W
W7-Aminoactinomycin D (7-AAD) is a fluorescent chemical compound with a strong affinity for DNA. It is used as a fluorescent marker for DNA in fluorescence microscopy and flow cytometry. It intercalates in double-stranded DNA, with a high affinity for GC-rich regions, making it useful for chromosome banding studies.
 W
WBrainbow is a process by which individual neurons in the brain can be distinguished from neighboring neurons using fluorescent proteins. By randomly expressing different ratios of red, green, and blue derivatives of green fluorescent protein in individual neurons, it is possible to flag each neuron with a distinctive color. This process has been a major contribution to the field of connectomics, traditionally known as hodology, which is the study of neural connections in the brain.
 W
WCellomics is the discipline of quantitative cell analysis using bioimaging methods and informatics with a workflow involving three major components: image acquisition, image analysis, and data visualization and management. These processes are generally automated. All three of these components depend on sophisticated software to acquire qualitative data, quantitative data, and the management of both images and data, respectively. Cellomics is also a trademarked term, which is often used interchangeably with high-content analysis (HCA) or high-content screening (HCS), but cellomics extends beyond HCA/HCS by incorporating sophisticated informatics tools.
 W
WConfocal microscopy, most frequently confocal laser scanning microscopy (CLSM) or laser confocal scanning microscopy (LCSM), is an optical imaging technique for increasing optical resolution and contrast of a micrograph by means of using a spatial pinhole to block out-of-focus light in image formation. Capturing multiple two-dimensional images at different depths in a sample enables the reconstruction of three-dimensional structures within an object. This technique is used extensively in the scientific and industrial communities and typical applications are in life sciences, semiconductor inspection and materials science.
 W
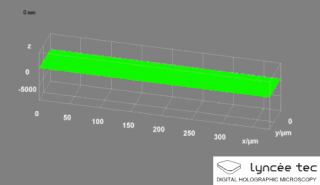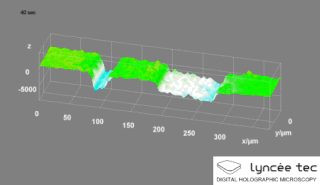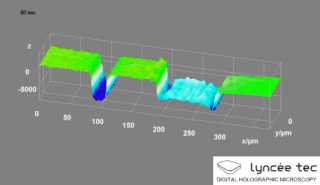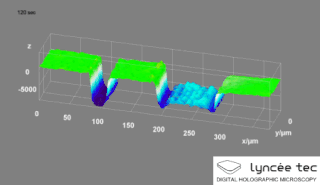
WDigital holographic microscopy (DHM) is digital holography applied to microscopy. Digital holographic microscopy distinguishes itself from other microscopy methods by not recording the projected image of the object. Instead, the light wave front information originating from the object is digitally recorded as a hologram, from which a computer calculates the object image by using a numerical reconstruction algorithm. The image forming lens in traditional microscopy is thus replaced by a computer algorithm. Other closely related microscopy methods to digital holographic microscopy are interferometric microscopy, optical coherence tomography and diffraction phase microscopy. Common to all methods is the use of a reference wave front to obtain amplitude (intensity) and phase information. The information is recorded on a digital image sensor or by a photodetector from which an image of the object is created (reconstructed) by a computer. In traditional microscopy, which do not use a reference wave front, only intensity information is recorded and essential information about the object is lost.
 W
WFluo-3 is a fluorescence indicator of intracellular calcium (Ca2+). It is used to measure Ca2+ inside living cells in flow cytometry and confocal laser scanning microscopy using visible light excitation (compatible with argon laser sources operating at 488 nm). Fluo-3 is an essentially nonfluorescent compound, but upon binding of Ca2+ its fluorescence increases sharply with an emission maximum at 525 nm suitable for conventionally used detectors designed for fluorescein isothiocyanate (FITC) measurements. This large change in fluorescence coupled with a good yield of photons provides very high contrast which allowed the detection of microscopic Ca2+ release events inside cells called "Calcium sparks". Whereas the salts of fluo-3 are unable to penetrate cells, loading can be achieved using its acetoxymethyl (AM) ester derivative. Once inside the cell, unspecific esterases cleave the ester effectively trapping fluo-3.
 W
WFluo-4 is used to measure calcium (Ca2+) concentrations inside living cells, and is often used for high-throughput screening of receptor ligands and calcium permeable ion channels.
 W
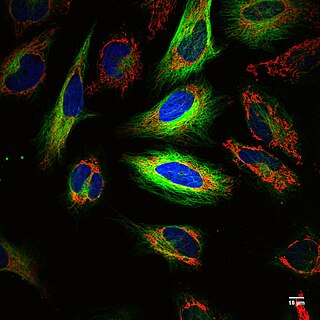
WFluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.
 W
WA fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a more simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.
 W
WFluorescence recovery after photobleaching (FRAP) is a method for determining the kinetics of diffusion through tissue or cells. It is capable of quantifying the two dimensional lateral diffusion of a molecularly thin film containing fluorescently labeled probes, or to examine single cells. This technique is very useful in biological studies of cell membrane diffusion and protein binding. In addition, surface deposition of a fluorescing phospholipid bilayer allows the characterization of hydrophilic surfaces in terms of surface structure and free energy.
 W
WA FMN-binding fluorescent protein (FbFP), also known as a LOV-based fluorescent protein, is a small, oxygen-independent fluorescent protein that binds flavin mononucleotide (FMN) as a chromophore.
 W
WFörster resonance energy transfer (FRET), fluorescence resonance energy transfer (FRET), resonance energy transfer (RET) or electronic energy transfer (EET) is a mechanism describing energy transfer between two light-sensitive molecules (chromophores). A donor chromophore, initially in its electronic excited state, may transfer energy to an acceptor chromophore through nonradiative dipole–dipole coupling. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making FRET extremely sensitive to small changes in distance.
 W
WFura-2, an aminopolycarboxylic acid, is a ratiometric fluorescent dye which binds to free intracellular calcium. It was the first widely used dye for calcium imaging, and remains very popular. Fura-2 is excited at 340 nm and 380 nm of light, and the ratio of the emissions at those wavelengths is directly related to the amount of intracellular calcium. Regardless of the presence of calcium, Fura-2 emits at 510 nm of light. The use of the ratio automatically cancels out confounding variables, such as variable dye concentration and cell thickness, making Fura-2 one of the most appreciated tools to quantify calcium levels. The high photon yield of fura-2 allowed the first real time measurements of calcium inside living cells in 1986. More recently, genetically-encoded calcium indicators based on spectral variants of the green fluorescent protein, such as Cameleons, have supplemented the use of Fura-2 and other small molecule dyes for calcium imaging, but Fura-2 remains faster.
 W
WFura-2-acetoxymethyl ester, often abbreviated Fura-2AM, is a membrane-permeant derivative of the ratiometric calcium indicator Fura-2 used in biochemistry to measure cellular calcium concentrations by fluorescence. When added to cells, Fura-2AM crosses cell membranes and once inside the cell, the acetoxymethyl groups are removed by cellular esterases. Removal of the acetoxymethyl esters regenerates "Fura-2", the pentacarboxylate calcium indicator. Measurement of Ca2+-induced fluorescence at both 340 nm and 380 nm allows for calculation of calcium concentrations based 340/380 ratios. The use of the ratio automatically cancels out certain variables such as local differences in fura-2 concentration or cell thickness that would otherwise lead to artifacts when attempting to image calcium concentrations in cells.
 W
WGCaMP is a genetically encoded calcium indicator, or GECI initially developed by Junichi Nakai. GCaMP is created from a fusion of green fluorescent protein (GFP), calmodulin, and M13, a peptide sequence from myosin light chain kinase. The advantage of GECI's is that they can be genetically specified for studies in living organisms. The first transgenic mouse expressing a GCaMP was reported in 2004 and GCaMP was subsequently improved to GCaMP2, which was stable at mammalian body temperatures and enabled the first in vivo mammalian recordings using a GECI. GCaMPs have been subsequently modified to progressively improve the range of the fluorescence signal, resulting in GCaMP3 through GCaMP-X. Additionally, red fluorescence GECIs, termed "RCaMPs" have been developed to expand the spectral options for multi-lineage imaging.
 W
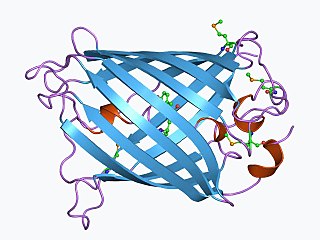
WThe green fluorescent protein (GFP) is a protein that exhibits bright green fluorescence when exposed to light in the blue to ultraviolet range. The label GFP traditionally refers to the protein first isolated from the jellyfish Aequorea victoria and is sometimes called avGFP. However, GFPs have been found in other organisms such as copepods and lancelets.
 W
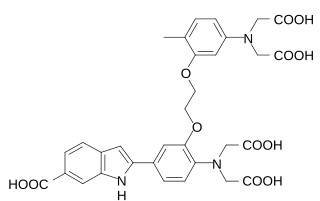
WIndo-1 is a popular calcium indicator similar to Fura-2. In contrast to Fura-2, Indo-1 has a dual emissions peak. The main emission peak in calcium-free solution is 475 nm while in the presence of calcium the emission is shifted to 400 nm. It is widely used in flow cytometry. The penta potassium salt is commercially available and preferred to the free acid because of its higher solubility in water. While Indo-1 is not cell permeable the penta acetoxymethyl ester Indo-1 AM enters the cell where it is cleaved by intracellular esterases to Indo-1. The synthesis and properties of Indo-1 were presented in 1985 by the group of Roger Y Tsien.
 W
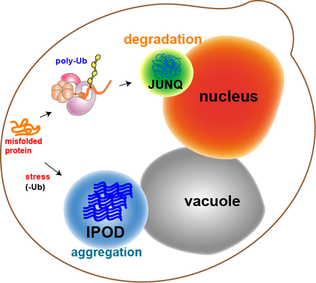
WJUNQ and IPOD are types of cytosolic protein inclusion bodies in eukaryotes.
 W
WLaser capture microdissection (LCM), also called microdissection, laser microdissection (LMD), or laser-assisted microdissection, is a method for isolating specific cells of interest from microscopic regions of tissue/cells/organisms.
 W
WLight sheet fluorescence microscopy (LSFM) is a fluorescence microscopy technique with an intermediate-to-high optical resolution, but good optical sectioning capabilities and high speed. In contrast to epifluorescence microscopy only a thin slice of the sample is illuminated perpendicularly to the direction of observation. For illumination, a laser light-sheet is used, i.e. a laser beam which is focused only in one direction. A second method uses a circular beam scanned in one direction to create the lightsheet. As only the actually observed section is illuminated, this method reduces the photodamage and stress induced on a living sample. Also the good optical sectioning capability reduces the background signal and thus creates images with higher contrast, comparable to confocal microscopy. Because LSFM scans samples by using a plane of light instead of a point, it can acquire images at speeds 100 to 1000 times faster than those offered by point-scanning methods.
 W
WLive cell imaging is the study of living cells using time-lapse microscopy. It is used by scientists to obtain a better understanding of biological function through the study of cellular dynamics. Live cell imaging was pioneered in first decade of the 20th century. One of the first time-lapse microcinematographic films of cells ever made was made by Julius Ries, showing the fertilization and development of the sea urchin egg. Since then, several microscopy methods have been developed which allow researchers to study living cells in greater detail with less effort. A newer type of imaging utilizing quantum dots have been used as they are shown to be more stable. The development of holotomographic microscopy has disregarded phototoxicity and other staining-derived disadvantages by implementing digital staining based on cells’ refractive index.
 W
WMicrographia: or Some Physiological Descriptions of Minute Bodies Made by Magnifying Glasses. With Observations and Inquiries Thereupon. is a historically significant book by Robert Hooke about his observations through various lenses. It is particularly notable for being the first book to illustrate insects, plants etc. as seen through microscopes. Published in January 1665, the first major publication of the Royal Society, it became the first scientific best-seller, inspiring a wide public interest in the new science of microscopy. It is also notable for coining the biological term cell.
 W
WMultifocal plane microscopy (MUM) or Multiplane microscopy or Biplane microscopy is a form of light microscopy that allows the tracking of the 3D dynamics in live cells at high temporal and spatial resolution by simultaneously imaging different focal planes within the specimen. In this methodology, the light collected from the sample by an infinity-corrected objective lens is split into two paths. In each path the split light is focused onto a detector which is placed at a specific calibrated distance from the tube lens. In this way, each detector images a distinct plane within the sample. The first developed MUM setup was capable of imaging two distinct planes within the sample. However, the setup can be modified to image more than two planes by further splitting the light in each light path and focusing it onto detectors placed at specific calibrated distances. Another technique called multifocus microscopy (MFM) uses diffractive Fourier optics to image up to 25 focal planes. Presently, MUM setups are implemented that can image up to four distinct planes.
 W
WPhasor approach refers to a method which is used for vectorial representation of sinusoidal waves like alternative currents and voltages or electromagnetic waves. The amplitude and the phase of the waveform is transformed into a vector where the phase is translated to the angle between the phasor vector and X axis and the amplitude is translated to vector length or magnitude. In this concept the representation and the analysis becomes very simple and the addition of two wave forms is realized by their vectorial summation.
 W
WIn optics, photobleaching is the photochemical alteration of a dye or a fluorophore molecule such that it permanently is unable to fluoresce. This is caused by cleaving of covalent bonds or non-specific reactions between the fluorophore and surrounding molecules. Such irreversible modifications in covalent bonds are caused by transition from a singlet state to the triplet state of the fluorophores. The number of excitation cycles to achieve full bleaching varies. In microscopy, photobleaching may complicate the observation of fluorescent molecules, since they will eventually be destroyed by the light exposure necessary to stimulate them into fluorescing. This is especially problematic in time-lapse microscopy.
 W
WIn biochemistry and molecular biology, a pulse-chase analysis is a method for examining a cellular process occurring over time by successively exposing the cells to a labeled compound (pulse) and then to the same compound in an unlabeled form (chase).
 W
WQuantitative phase contrast microscopy or quantitative phase imaging are the collective names for a group of microscopy methods that quantify the phase shift that occurs when light waves pass through a more optically dense object.
 W
WSecond-harmonic imaging microscopy (SHIM) is based on a nonlinear optical effect known as second-harmonic generation (SHG). SHIM has been established as a viable microscope imaging contrast mechanism for visualization of cell and tissue structure and function. A second-harmonic microscope obtains contrasts from variations in a specimen's ability to generate second-harmonic light from the incident light while a conventional optical microscope obtains its contrast by detecting variations in optical density, path length, or refractive index of the specimen. SHG requires intense laser light passing through a material with a noncentrosymmetric molecular structure. Second-harmonic light emerging from an SHG material is exactly half the wavelength (frequency doubled) of the light entering the material. While two-photon-excited fluorescence (TPEF) is also a two photon process, TPEF loses some energy during the relaxation of the excited state, while SHG is energy conserving. Typically, an inorganic crystal is used to produce SHG light such as lithium niobate (LiNbO3), potassium titanyl phosphate (KTP = KTiOPO4), and lithium triborate (LBO = LiB3O5). Though SHG requires a material to have specific molecular orientation in order for the incident light to be frequency doubled, some biological materials can be highly polarizable, and assemble into fairly ordered, large noncentrosymmetric structures. Biological materials such as collagen, microtubules, and muscle myosin can produce SHG signals. The SHG pattern is mainly determined by the phase matching condition. A common setup for an SHG imaging system will have a laser scanning microscope with a titanium sapphire mode-locked laser as the excitation source. The SHG signal is propagated in the forward direction. However, some experiments have shown that objects on the order of about a tenth of the wavelength of the SHG produced signal will produce nearly equal forward and backward signals.
 W
WThe need for fluorescently tracking RNA rose as its roles in complex cellular functions has grown to not only include mRNA, rRNA, and tRNA, but also RNAi, siRNA, snoRNA, and lncRNA, among others. Spinach is a synthetically derived RNA aptamer born out of the need for a way of studying the role of RNAs at the cellular level. This aptamer was created using Systematic Evolution for Ligands by EXponential enrichment, or SELEX, which is also known as in vitro evolution.
WTime-lapse microscopy is time-lapse photography applied to microscopy. Microscope image sequences are recorded and then viewed at a greater speed to give an accelerated view of the microscopic process.
 W
WA total internal reflection fluorescence microscope (TIRFM) is a type of microscope with which a thin region of a specimen, usually less than 200 nanometers can be observed.
 W
WTwo-photon excitation microscopy is a fluorescence imaging technique that allows imaging of living tissue up to about one millimeter in thickness. Unlike traditional fluorescence microscopy, in which the excitation wavelength is shorter than the emission wavelength, two-photon excitation requires simultaneous excitation by two photons with longer wavelength than the emitted light. Two-photon excitation microscopy typically uses near-infrared (NIR) excitation light which can also excite fluorescent dyes. However, for each excitation, two photons of NIR light are absorbed. Using infrared light minimizes scattering in the tissue. Due to the multiphoton absorption, the background signal is strongly suppressed. Both effects lead to an increased penetration depth for this technique. Two-photon excitation can be a superior alternative to confocal microscopy due to its deeper tissue penetration, efficient light detection, and reduced photobleaching.